本节主要介绍使用VASP计算时,常用的一些工具。
VASPKIT VASPKIT 是一个针对VASP的程序集,通过命令行调用可以方便地生成与修改输入文件 和初步处理输出数据 ,一些常用的命令如下:
Command-预处理
Function
vaspkit -task 102
生成KPOINTS
vaspkit -task 103
生成POTCAR,默认PBE
vaspkit -task 105
从cif文件生成POSCAR
vaspkit -task 303
生成KPOINTS,用于体相能带计算
vaspkit -task 601
给出晶体对称性
vaspkit -task 602
给出原胞的POSCAR
vaspkit -task 603
给出惯用晶胞的POSCAR
vaspkit -task 604
给出对称等价的原子
vaspkit -task 608
给出弛豫结构的对称性
Command-后处理 Function
vaspkit -task 111
提取总的态密度
vaspkit -task 113
提取每种元素的投影态密度
vaspkit -task 211
提取能带
vaspkit -task 213
提取每种元素的投影能带
vaspkit -task 263
FermiSurfer格式的费米面
Python python用于提取数据与可视化结果。
Vesta Vesta用于可视化和编辑晶体结构文件。
p4vasp P4VASP通过读取vasprun.xml文件,便捷地可视化输出文件。
1 Examples Reproduce 1.1 Bulk Systems 1.1.1 fcc Si 任务 找到 fcc Si 的最佳晶格参数。
输入文件 1 2 3 4 5 6 7 8 9 10 ##POSCAR fcc Si: 3.9 0.5 0.5 0.0 0.0 0.5 0.5 0.5 0.0 0.5 1 Si cartesian 0 0 0
1 2 3 4 5 ##INCAR System = fcc Si ISTART = 0 ; ICHARG = 2 ENCUT = 500 ISMEAR = 0; SIGMA = 0.1
自行准备KPOINTS, POTCAR。
计算
计算不同晶格参数的能量。
拟合某些状态方程获得平衡体积。(可尝试vaspkit)
bash脚本loop.sh计算不同晶格常数(3.5-4.3$\mathring{A}$)的fcc Si并提取对应的自由能于SUMMARY.fcc中。
注意-在VASP6.5.1中,ENCUT增加至约500才能使得脚本中的所有晶格参数所对应的计算收敛。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 rm WAVECAR SUMMARY.fccfor i in 3.5 3.6 3.7 3.8 3.9 4.0 4.1 4.2 4.3 ; do cat >POSCAR <<!fcc: $i 0.5 0.5 0.0 0.0 0.5 0.5 0.5 0.0 0.5 1 cartesian 0 0 0 ! echo "a= $i " mpirun vasp_std &> vasp.log E=`awk '/F=/ {print $0}' OSZICAR` ; echo $i $E >>SUMMARY.fcc done cat SUMMARY.fcc
1 2 3 4 5 6 7 8 9 10 ##SUMMARY.fcc 3.5 1 F= -.44610629E+01 E0= -.44588862E+01 d E =-.435335E-02 3.6 1 F= -.46872871E+01 E0= -.46859153E+01 d E =-.274377E-02 3.7 1 F= -.48211455E+01 E0= -.48190962E+01 d E =-.409845E-02 3.8 1 F= -.48862039E+01 E0= -.48846919E+01 d E =-.302386E-02 3.9 1 F= -.48966342E+01 E0= -.48951129E+01 d E =-.304247E-02 4.0 1 F= -.48652835E+01 E0= -.48646264E+01 d E =-.131428E-02 4.1 1 F= -.47993069E+01 E0= -.47985430E+01 d E =-.152769E-02 4.2 1 F= -.47047953E+01 E0= -.47033649E+01 d E =-.286075E-02 4.3 1 F= -.45911248E+01 E0= -.45891970E+01 d E =-.385564E-02
1.1.2 fcc Si DOS 任务 计算fcc Si的态密度
输入文件 1 2 3 4 5 6 7 8 9 10 ##POSCAR fcc Si: 3.9 0.5 0.5 0.0 0.0 0.5 0.5 0.5 0.0 0.5 1 Si cartesian 0 0 0
1 2 3 4 5 6 7 8 9 10 11 ##INCAR System = fcc Si ICHARG = 11 # read CHGCAR file ENCUT = 500 ISMEAR = -5 # tetrahedron方法 LORBIT = 11 # projection ##E-range and density #Emin = #Emax = #NEDOS =
自行准备KPOINTS, POTCAR。
计算
使用下面的脚本从scf文件夹中复制DOS计算所需文件并提交任务。
1 2 3 4 5 6 7 8 9 10 11 mkdir doscp incar.2.dos dos/INCAR for ii in POSCAR POTCAR KPOINTS CHGCAR;do cp scf/${ii} dosdone cd dosawk 'NR==4 {$1=$1*2+1;$2=$2*2+1;$3=$3*2+1} 1' KPOINTS > KPOINTS_dense && cp KPOINTS_dense KPOINTS mpirun vasp_std &> dos.log
对大的体系:
先用较少的k点收敛。
计算DOS时增加k点密度并通过设置ICHARG = 11,使电荷密度与电势能固定。
设置ISMEAR = -5,使用blochl修正的四面体积分方法。
使用p4vasp或python绘制DOS。
使用下面的bash脚本调用awk 与gnuplot 来绘制dos。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 awk 'BEGIN{i=1} /dos>/,\ /\/dos>/ \ {a[i]=$2 ; b[i]=$3 ; i=i+1} \ END{for (j=12;j<i-5;j++) print a[j],b[j]}' vasprun.xml > dos.datef=`awk '/efermi/ {print $3}' vasprun.xml` cat >plotfile<<!plot "dos.dat" using (\$1 -$ef ):(\$2 ) w lp ! gnuplot -persist plotfile rm dos.dat plotfile
1.1.3 fcc Si bandstructure 任务 计算fcc Si能带。
输入文件 1 2 3 4 5 6 7 8 9 10 ##POSCAR fcc Si: 3.9 0.5 0.5 0.0 0.0 0.5 0.5 0.5 0.0 0.5 1 Si cartesian 0 0 0
1 2 3 4 5 6 ##INCAR System = fcc Si ICHARG = 11 # read CHGCAR file ENCUT = 500 ISMEAR = 0; SIGMA = 0.1 LORBIT = 10 # 10 for l-decomposed and 11 for lm-decomposed
使用vaspkit自动生成或手动写入高对称k点。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 ##KPOINTS k-points for bandstructure L-G-X-U K-G 10 liciprocal kkaokaokaokaokaokaokaokaokaokaone reciprocal 0.50000 0.50000 0.50000 1 0.00000 0.00000 0.00000 1 0.00000 0.00000 0.00000 1 0.00000 0.50000 0.50000 1 0.00000 0.50000 0.50000 1 0.25000 0.62500 0.62500 1 0.37500 0.7500 0.37500 1 0.00000 0.00000 0.00000 1
计算
使用下面的脚本从scf中复制BAND计算所需文件。
1 2 3 4 5 6 7 8 9 10 11 12 mkdir bandcp incar.3.band band/INCAR for ii in POSCAR POTCAR CHGCAR;do cp scf/${ii} banddone cd bandvaspkit -task 303 cp KPATH.in KPOINTSmpirun vasp_std &> band.log
1.1.4 cd Si relaxation 任务 弛豫cubic diamond Si的内部坐标,体积与单胞形状。
输入文件 1 2 3 4 5 6 7 8 9 10 11 ##POSCAR cubic diamond 5.5 0.0 0.5 0.5 0.5 0.0 0.5 0.5 0.5 0.0 Si 2 Direct -0.125 -0.125 -0.125 0.125 0.125 0.125
1 2 3 4 5 6 7 8 ##INCAR System = diamond Si ISMEAR = 0; SIGMA = 0.1; ENMAX = 500 IBRION = 2; ISIF=3 ; NSW=100 PREC = high #set the FFT grids, the accuracy of the projectors in real space. EDIFF = 0.1E-06 #EDIFFG = -0.01 #default is equal to EDIFF * 10
自行准备KPOINTS, POTCAR
计算 1.1.5 fcc Ni 任务 计算自旋极化(ISPIN = 2)的fcc Ni。
输入文件 1 2 3 4 5 6 7 8 9 10 ##POSCAR fcc: 3.53 0.5 0.5 0.0 0.0 0.5 0.5 0.5 0.0 0.5 Ni 1 direct 0 0 0
1 2 3 4 5 6 7 8 ##INCAR SYSTEM = fcc Ni ISTART = 0 ; ICHARG=2 ENCUT = 500 ISMEAR = 1 ; SIGMA = 0.2 EDIFF = 1E-06 ISPIN = 2; ISYM = -1 MAGMOM = 1
自行生成KPOINTS,POTCAR
计算 1.1.6 Graphite TS binding energy 任务 使用Tchatchenko-Scheffler方法确定实验结构石墨的层间结合能。
输入文件 1 2 3 4 5 6 7 8 9 10 11 12 ##POSCAR graphite 1.0 1.22800000 -2.12695839 0.00000000 1.22800000 2.12695839 0.00000000 0.00000000 0.00000000 6.71 4 direct 0.00000000 0.00000000 0.25000000 0.00000000 0.00000000 0.75000000 0.33333333 0.66666667 0.25000000 0.66666667 0.33333333 0.75000000
1 2 3 4 5 6 ##KPOINTS K-Spacing Value to Generate K-Mesh: 0.040 0 Gamma 15 15 4 0.0 0.0 0.0
1 2 3 4 5 6 7 8 9 10 11 ##POSCAR graphene 1.0 1.22800000 -2.12695839 0.00000000 1.22800000 2.12695839 0.00000000 0.00000000 0.00000000 20. C 2 direct 0.00000000 0.00000000 0.25000000 0.33333333 0.66666667 0.25000000
1 2 3 4 5 6 ##KPOINTS K-Spacing Value to Generate K-Mesh: 0.040 0 Gamma 15 15 1 0.0 0.0 0.0
1 2 3 4 5 6 7 8 9 ##INCAR System = graphite ISMEAR = 0; SIGMA = 0.01 ENCUT = 500 LWAVE = .FALSE. LCHARG = .FALSE. EDIFF = 1e-6 IVDW = 20 LVDW_EWALD =.TRUE.
计算 分别提交graphite和graphene的计算,使用下面的脚本提取结合能。
1 2 3 4 5 6 en2=$(grep "free ene" graphite/OUTCAR |tail -1|awk '{print $5}' ) en1=$(grep "free ene" graphene/OUTCAR |tail -1|awk '{print $5}' ) deltaE=$(echo "$en2 /4 - $en1 /2" | bc -l) echo "Binding energy (eV/atom):" $deltaE > results.dat
1.1.7 Graphite MBD binding energy 任务 使用MBD方法计算实验结构石墨的层间结合能。
输入文件 POSCAR和KPOINTS与1.1.6-TS方法中相同。
1 2 3 4 5 6 7 8 9 ##INCAR System = graphite ISMEAR = 0; SIGMA = 0.01 ENCUT = 500 LWAVE = .FALSE. LCHARG = .FALSE. EDIFF = 1e-6 IVDW = 202 LVDWEXPANSION = .TRUE.
计算 分别提交graphite和graphene的计算,使用下面的脚本提取结合能。
1 2 3 4 5 6 en2=$(grep "free ene" graphite/OUTCAR |tail -1|awk '{print $5}' ) en1=$(grep "free ene" graphene/OUTCAR |tail -1|awk '{print $5}' ) deltaE=$(echo "$en2 /4 - $en1 /2" | bc -l) echo "Binding energy (eV/atom):" $deltaE > results.dat
1.2 Calculate $U$ for LSDA+$U$ 任务 输入文件 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 ##POSCAR AFM NiO 4.03500000 2.0000000000 1.0000000000 1.0000000000 1.0000000000 2.0000000000 1.0000000000 1.0000000000 1.0000000000 2.0000000000 1 15 16 Direct 0.0000000000 0.0000000000 0.0000000000 0.2500000000 0.2500000000 0.2500000000 0.0000000000 0.0000000000 0.5000000000 0.2500000000 0.2500000000 0.7500000000 0.0000000000 0.5000000000 0.0000000000 0.2500000000 0.7500000000 0.2500000000 0.0000000000 0.5000000000 0.5000000000 0.2500000000 0.7500000000 0.7500000000 0.5000000000 0.0000000000 0.0000000000 0.7500000000 0.2500000000 0.2500000000 0.5000000000 0.0000000000 0.5000000000 0.7500000000 0.2500000000 0.7500000000 0.5000000000 0.5000000000 0.0000000000 0.7500000000 0.7500000000 0.2500000000 0.5000000000 0.5000000000 0.5000000000 0.7500000000 0.7500000000 0.7500000000 0.1250000000 0.1250000000 0.1250000000 0.3750000000 0.3750000000 0.3750000000 0.1250000000 0.1250000000 0.6250000000 0.3750000000 0.3750000000 0.8750000000 0.1250000000 0.6250000000 0.1250000000 0.3750000000 0.8750000000 0.3750000000 0.1250000000 0.6250000000 0.6250000000 0.3750000000 0.8750000000 0.8750000000 0.6250000000 0.1250000000 0.1250000000 0.8750000000 0.3750000000 0.3750000000 0.6250000000 0.1250000000 0.6250000000 0.8750000000 0.3750000000 0.8750000000 0.6250000000 0.6250000000 0.1250000000 0.8750000000 0.8750000000 0.3750000000 0.6250000000 0.6250000000 0.6250000000 0.8750000000 0.8750000000 0.8750000000
1 2 3 4 5 6 ##KPOINTS Gamma only 0 Monkhorst 1 1 1 0 0 0
计算 1.3 Magnetism 1.2.3 NiO LSDA+$U$ 任务 使用DFT+$U$(Dudarev方法)计算反铁磁 NiO。
输入文件 1 2 3 4 5 6 7 8 9 10 11 12 ##POSCAR AFM NiO 4.17 1.0 0.5 0.5 0.5 1.0 0.5 0.5 0.5 1.0 2 2 Cartesian 0.0 0.0 0.0 1.0 1.0 1.0 0.5 0.5 0.5 1.5 1.5 1.5
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 ##INCAR SYSTEM = NiO ISTART = 0 ISPIN = 2 MAGMOM = 2.0 -2.0 2*0 ENMAX = 250.0 EDIFF = 1E-3 ISMEAR = -5 AMIX = 0.2 BMIX = 0.00001 AMIX_MAG = 0.8 BMIX_MAG = 0.00001 LORBIT = 11 LDAU = .TRUE. LDAUTYPE = 2 LDAUL = 2 -1 LDAUU = 8.00 0.00 LDAUJ = 0.95 0.00 LDAUPRINT = 1 LMAXMIX = 4 ! Important: mix paw occupancies up to L=4
1 2 3 4 5 6 ##KPOINTS k-points 0 gamma 4 4 4 0 0 0
计算 1.4 BSE 1.4.1 Dielectric properties of Si using BSE 任务 通过求解GW0上的Bethe-Salpeter方程(BSE),计算包含激子效应的Si的介电函数。
输入文件 1 2 3 4 5 6 7 8 9 10 11 ##POSCAR Si 5.4300 0.5 0.5 0.0 0.0 0.5 0.5 0.5 0.0 0.5 Si 2 cart 0.00 0.00 0.00 0.25 0.25 0.25
1 2 3 4 5 6 ##KPOINTS K-Spacing Value to Generate K-Mesh: 0.040 0 Gamma 6 6 6 0.0 0.0 0.0
自行准备POTCAR文件。
1 2 3 4 5 ##incar.1.dft System = Si ENCUT = 500 ISMEAR = 0 ; SIGMA = 0.01 EDIFF = 1.E-8
1 2 3 4 5 6 7 8 ##incar.2.diag System = Si PREC = Normal ; ENCUT = 500 ALGO = EXACT ; NELM = 1 ISMEAR = 0 ; SIGMA = 0.01 NBANDS = 128 LOPTICS = .TRUE. ; LPEAD = .TRUE. OMEGAMAX = 40
1 2 3 4 5 6 7 8 9 10 11 ##incar.3.gw0 System = Si PREC = Normal ; ENCUT = 500 ALGO = GW0 ISMEAR = 0 ; SIGMA = 0.01 ENCUTGW = 150 ; NELM = 1 ; NOMEGA = 50 ; OMEGATL = 280 #NBANDSO=4 ; NBANDSV=8 ; LADDER=.TRUE. ; LUSEW=.TRUE. NBANDS = 128 NBANDSGW = 12 LWAVE = .TRUE. PRECFOCK = Normal
1 2 3 4 5 6 7 8 9 10 ##incar.4.none System = Si PREC = Normal ; ENCUT = 500 ALGO = Nothing ; NELM = 1 ISMEAR = 0 ; SIGMA = 0.01 KPAR = 2 NBANDS = 128 LWAVE = .FALSE. LOPTICS = .TRUE. ; LPEAD = .TRUE. OMEGAMAX = 40
1 2 3 4 5 6 7 8 9 10 11 12 13 ##incar.5.bse SYSTEM = Si PREC = Normal ; ENCUT = 500 ALGO = BSE ANTIRES = 0 ISMEAR = 0 ; SIGMA = 0.01 ENCUTGW = 150 EDIFF = 1.E-8 NBANDS = 128 NBANDSO = 4 NBANDSV = 8 OMEGAMAX = 20 PRECFOCK = Normal
计算 GW0+BSE计算的工作流程在doall.sh中给出,包含以下连续步骤:
标准的DFT基态计算
获取虚能带:需要步骤1中的WAVECAR。
GW0计算:需要步骤2中的WAVECAR和WAVEDER。
可选步骤:使用LOPTICS=.TRUE.来给出独立粒子近似(IPA)下的介电函数,使用GW0准粒子能量而非DFT能量。
BSE计算:需要步骤3中的WAVECAR和步骤2中的WAVEDER。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 rm WAVECAR* WAVEDER*cp incar.1.dft INCARmpirun vasp_std &> DFT.log cp OUTCAR OUTCAR.DFTcp vasprun.xml vasprun.DFT.xmlcp incar.2.diag INCARmpirun vasp_std &> DIAG.log cp OUTCAR OUTCAR.DIAGcp vasprun.xml vasprun.DIAG.xml./extract_optics.sh mv optics.dat optics.DFT.datcp incar.3.gwp INCARmpirun vasp_std &> GW0.log cp OUTCAR OUTCAR.GW0cp vasprun.xml vasprun.GW0.xmlcp incar.4.none INCARmpirun vasp_std &> NONE.log cp OUTCAR OUTCAR.NONEcp vasprun.xml vasprun.NONE.xml./extract_optics.sh mv optics.dat optics.RPA.datcp incar.5.bse INCARmpirun vasp_std &> BSE.log cp OUTCAR OUTCAR.BSEcp vasprun.xml vasprun.BSE.xml./extract_optics.sh mv optics.dat optics.BSE.dat
1 2 3 4 5 6 7 8 awk 'BEGIN{i=0} /<dielectricfunction>/,\ /<\/dielectricfunction>/ \ {if ($1=="<r>") {a[i]=$2 ; b[i]=($3+$4+$5)/3 ; c[i]=$4 ; d[i]=$5 ; i=i+1}} \ END{for (j=0;j<i/2;j++) print a[j],b[j],b[j+i/2]}' vasprun.xml > optics.dat
所有计算完成后,使用脚本plotall.sh绘制IPA和BSE下的吸收系数。
1 2 3 4 5 6 7 8 9 10 11 12 13 ##plotall.sh cat >plotfile<<! # set term postscript enhanced eps colour lw 2 "Helvetica" 20 # set output "optics.eps" set xrange [0:10] plot "optics.DFT.dat" using (\$1):(\$2) w l lt -1 lw 1 lc -1 title "DFT", \ "optics.RPA.dat" using (\$1):(\$2) w l lt -1 lw 1 lc 1 title "RPA", \ "optics.BSE.dat" using (\$1):(\$2) w l lt -1 lw 1 lc 3 title "BSE" ! gnuplot -persist plotfile
1.5 Molecular Dynamics MD计算常用NVT ensemble,这与许多真实的实验条件对应。ISIF=2;MDALGO=2;系综和thermostat通过这两个参数共同确定,该设置对应的是Canonical ensemble,Nose-Hoover thermostat。通过引入一个虚变量来代表环境的自由度,动力学地调节温度 。系统通过与这个虚变量交换能量来实现温度的恒定。
将系统的原子与一个假想的“热浴”耦合。
这个热浴会对原子施加一个摩擦力 。
当系统的瞬时温度高于 目标温度时,摩擦力会减慢 原子运动,从而降低动能和温度。
当系统的瞬时温度低于 目标温度时,摩擦力会加速 原子运动,从而增加动能和提高温度。
这个摩擦力的大小不是固定的,而是由一个额外的微分方程控制,该方程取决于当前温度与目标温度的差值。
1.5.1 Liquid Si - Standard MD 任务 通过分子动力学熔化晶体结构来生成液态Si。
输入文件 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 ##poscar111 Si cubic diamond conventional cell 5.43100000000000 1.00000000 0.00000000 0.00000000 0.00000000 1.00000000 0.00000000 0.00000000 0.00000000 1.00000000 Si 8 Direct 0.00000000 0.00000000 0.00000000 0.75000000 0.25000000 0.75000000 0.00000000 0.50000000 0.50000000 0.75000000 0.75000000 0.25000000 0.50000000 0.00000000 0.50000000 0.25000000 0.25000000 0.25000000 0.50000000 0.50000000 0.00000000 0.25000000 0.75000000 0.75000000
supercell $2\times2\times2$
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 ##poscar222 Si cubic diamond 2x2x2 super cell of conventional cell 5.43090000000000 2.00000000 0.00000000 0.00000000 0.00000000 2.00000000 0.00000000 0.00000000 0.00000000 2.00000000 Si 64 Direct 0.00000000 0.00000000 0.00000000 0.50000000 0.00000000 0.00000000 0.00000000 0.50000000 0.00000000 0.50000000 0.50000000 0.00000000 0.00000000 0.00000000 0.50000000 0.50000000 0.00000000 0.50000000 0.00000000 0.50000000 0.50000000 0.50000000 0.50000000 0.50000000 0.37500000 0.12500000 0.37500000 0.87500000 0.12500000 0.37500000 0.37500000 0.62500000 0.37500000 0.87500000 0.62500000 0.37500000 0.37500000 0.12500000 0.87500000 0.87500000 0.12500000 0.87500000 0.37500000 0.62500000 0.87500000 0.87500000 0.62500000 0.87500000 0.00000000 0.25000000 0.25000000 0.50000000 0.25000000 0.25000000 0.00000000 0.75000000 0.25000000 0.50000000 0.75000000 0.25000000 0.00000000 0.25000000 0.75000000 0.50000000 0.25000000 0.75000000 0.00000000 0.75000000 0.75000000 0.50000000 0.75000000 0.75000000 0.37500000 0.37500000 0.12500000 0.87500000 0.37500000 0.12500000 0.37500000 0.87500000 0.12500000 0.87500000 0.87500000 0.12500000 0.37500000 0.37500000 0.62500000 0.87500000 0.37500000 0.62500000 0.37500000 0.87500000 0.62500000 0.87500000 0.87500000 0.62500000 0.25000000 0.00000000 0.25000000 0.75000000 0.00000000 0.25000000 0.25000000 0.50000000 0.25000000 0.75000000 0.50000000 0.25000000 0.25000000 0.00000000 0.75000000 0.75000000 0.00000000 0.75000000 0.25000000 0.50000000 0.75000000 0.75000000 0.50000000 0.75000000 0.12500000 0.12500000 0.12500000 0.62500000 0.12500000 0.12500000 0.12500000 0.62500000 0.12500000 0.62500000 0.62500000 0.12500000 0.12500000 0.12500000 0.62500000 0.62500000 0.12500000 0.62500000 0.12500000 0.62500000 0.62500000 0.62500000 0.62500000 0.62500000 0.25000000 0.25000000 0.00000000 0.75000000 0.25000000 0.00000000 0.25000000 0.75000000 0.00000000 0.75000000 0.75000000 0.00000000 0.25000000 0.25000000 0.50000000 0.75000000 0.25000000 0.50000000 0.25000000 0.75000000 0.50000000 0.75000000 0.75000000 0.50000000 0.12500000 0.37500000 0.37500000 0.62500000 0.37500000 0.37500000 0.12500000 0.87500000 0.37500000 0.62500000 0.87500000 0.37500000 0.12500000 0.37500000 0.87500000 0.62500000 0.37500000 0.87500000 0.12500000 0.87500000 0.87500000 0.62500000 0.87500000 0.87500000
1 2 3 4 5 6 ##KPOINTS K-Points 0 Gamma 1 1 1 0 0 0
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 ##INCAR System = Si_MD IBRION = 0 MDALGO = 2 ISIF = 2 SMASS = 1.0 ISMEAR = 0; SIGMA = 0.1 LREAL = Auto ALGO = VeryFast PREC = Low ISYM = 0 TEBEG = 2000 NSW = 50 POTIM = 3.0 NCORE = 2
计算 对于超胞体计算,其第一布里渊区足够小,我们只对Gamma点进行抽样,使用程序vasp_gam进行计算。
1 2 awk <PCDAT >PCDAT.150fs ' NR==8 {pcskal=$1} NR==9 {pcfein=$1} NR>=13 {line=line+1; print (line-0.5)*pcfein/pcskal,$1} '
绘制pcf图像:
1 2 grep "free energy" OUTCAR|awk 'line=line+1;{print $5}' > 150fs.ene
绘制ene图像![[Si_ene.png]]
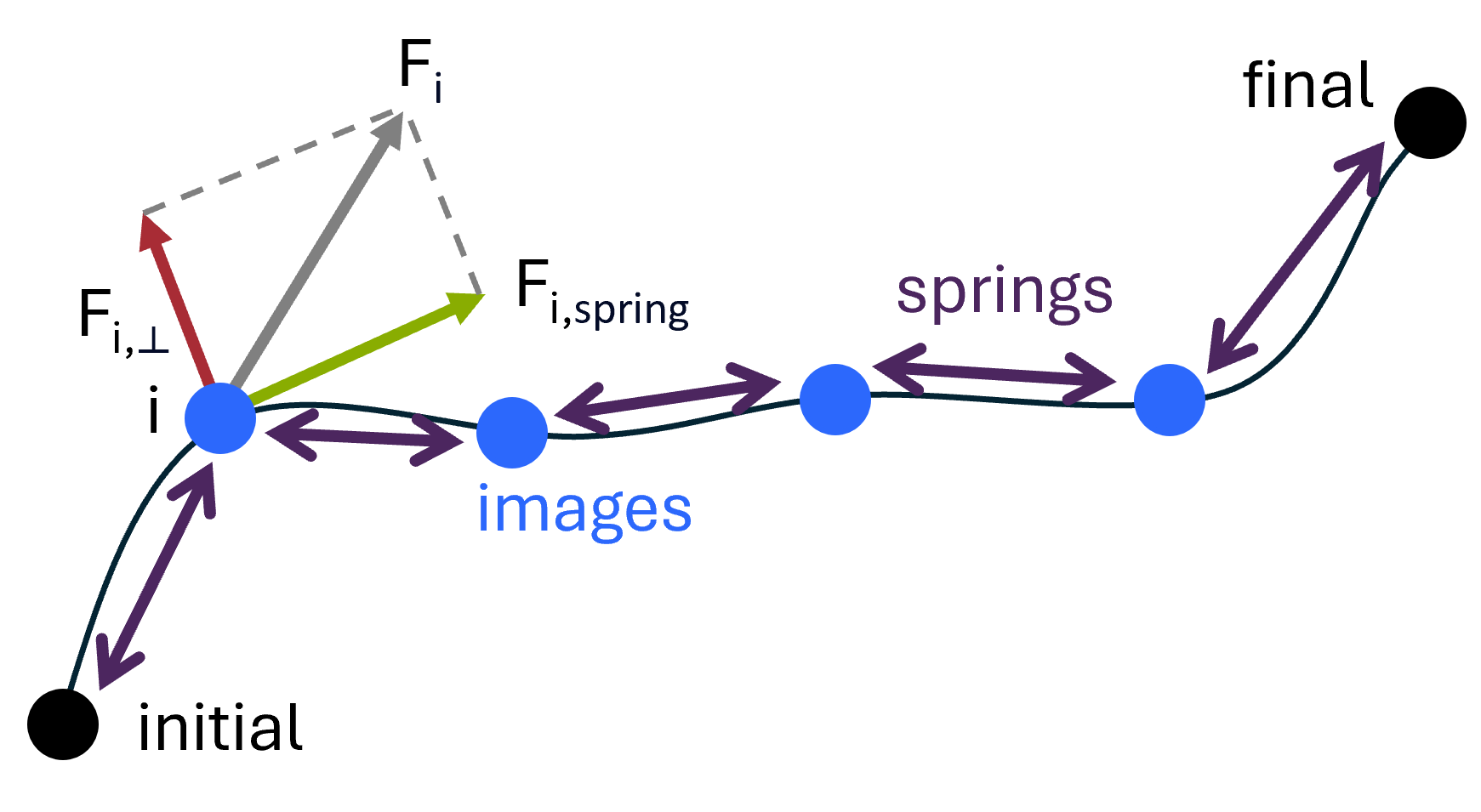
1.6 Transition states 在所有的化学反应中,从反应物到产物都会经历一个过渡态,这个过渡态是势能面上的鞍点。Nudged elastic band方法通过在反应物和产物间进行插值,来模拟最小能量路径。
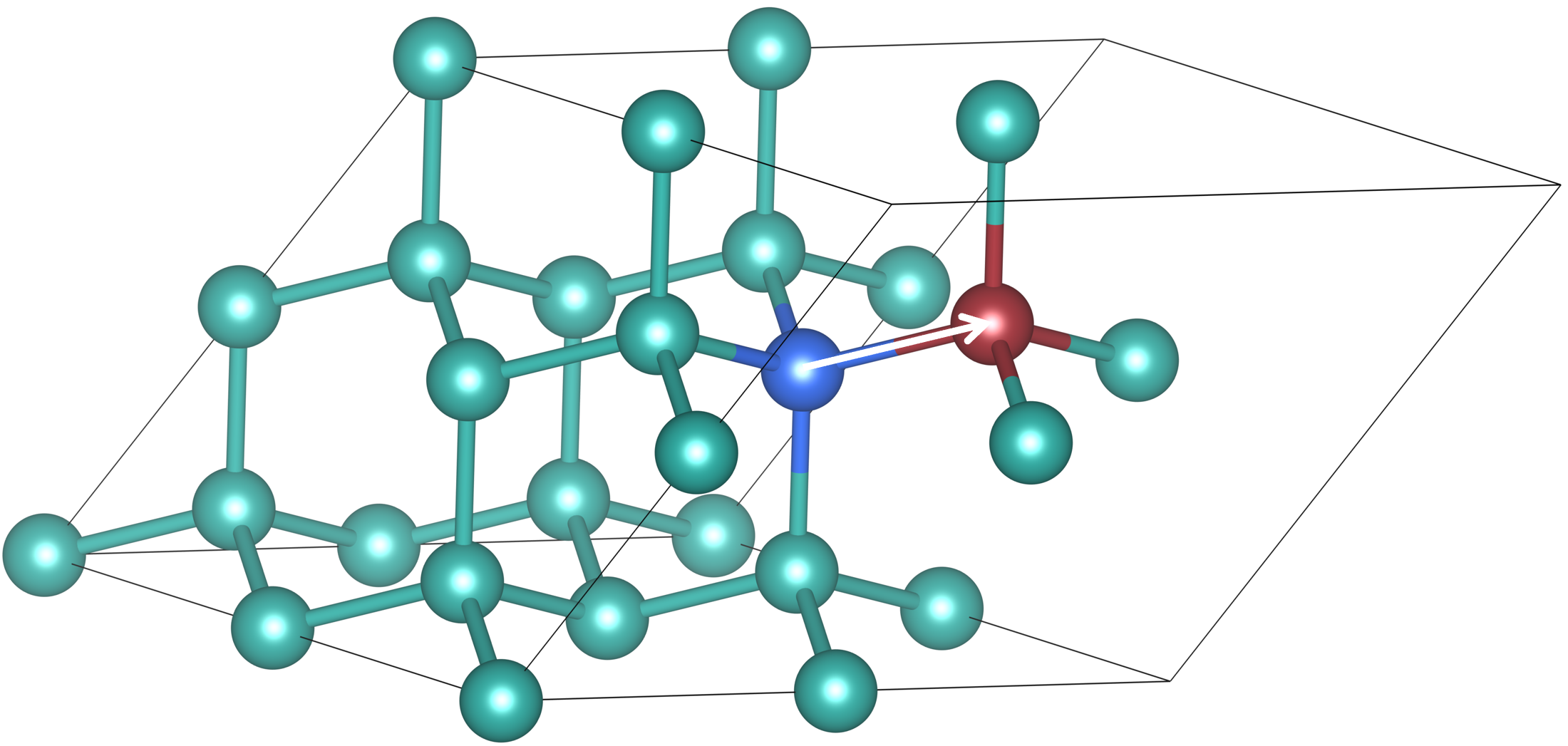
1.6.1 Reaction path with nudged elastic band on Si-self-diffusion 任务 使用NEB方法计算Si原子自扩散^[Phys. Rev. B 59, 3969 (1999) ]至15个Si原子的原始超胞中空位的反应路径。Si原子会从蓝色位置移动到红色的vacancy。
输入文件 Tutorial on how to generate images’ structure
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 from ase.io.vasp import read_vaspfrom ase.io.vasp import write_vaspfrom ase.mep.neb import NEBinitial = read_vasp("./POSCAR.V1" ) final = read_vasp("./POSCAR.V2" ) no_images = 4 images = [initial] images += [initial.copy() for i in range (no_images)] images += [final] neb = NEB(images) neb.interpolate(method="idpp" , apply_constraint=True ) a=0 for image in images: a += 1 write_vasp(("./e01_NEB/0" +str (a-1 )+"/POSCAR" ), image) if a == no_images+2 : break
准备00-06共六个文件夹,分别存放初态-4个中间态-末态的结构文件。
1 2 3 4 5 6 7 8 9 10 11 12 13 ##incar.sp System = Si ! Electronic minimization ENCUT = 250 # Plane-wave energy cutoff (eV) PREC = Normal # Precision tag EDIFF = 1e-6 # Break condition for the global, electronic SC step ! Smearing ISMEAR = 0 # Gaussian smearing SIGMA = 0.05 # Smearing width ! Single point NSW = 0 # Number of maximum ionic steps
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 ##incar.neb System = Si ! Electronic minimization ENCUT = 250 # Plane-wave energy cutoff (eV) PREC = Normal # Precision tag EDIFF = 1e-6 # Break condition for the global, electronic SC step ! Smearing ISMEAR = 0 # Gaussian smearing SIGMA = 0.05 # Smearing width ! Ionic relaxation NSW = 100 # Max number of ionic steps EDIFFG = -0.04 # Break condition for ionic relaxation IBRION = 1 # Quasi-Newton algorithm POTIM = 0.8 # Step width ISIF = 2 # Cell shape and volume fixed, ions free ! NEB IMAGES = 4 # 4 intermediate geometries for the NEB SPRING = -5 # Spring constant
cp incar.neb INCAR进行NEB计算;cp incar.sp 00/INCAR 05/INCAR进行初末态计算。
计算 在完成计算后,我们可以对原子沿着images的变化进行可视化:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 import matplotlib.pyplot as pltimport matplotlib.patches as mpatchesfrom ase.visualize.plot import plot_atomsfrom ase.io.vasp import read_vaspSi1 = read_vasp("./e01_NEB/00/CONTCAR" ) Si2 = read_vasp("./e01_NEB/01/CONTCAR" ) Si3 = read_vasp("./e01_NEB/02/CONTCAR" ) Si4 = read_vasp("./e01_NEB/03/CONTCAR" ) Si5 = read_vasp("./e01_NEB/04/CONTCAR" ) Si6 = read_vasp("./e01_NEB/05/CONTCAR" ) fig, ax = plt.subplots() xyz1 = '90x,45y,0z' plot_atoms(Si1, ax, radii=0.3 , rotation=(xyz1), colors = ["red" ]*15 ) plot_atoms(Si2, ax, radii=0.3 , rotation=(xyz1), colors = ["orange" ]*15 ) plot_atoms(Si3, ax, radii=0.3 , rotation=(xyz1), colors = ["yellow" ]*15 ) plot_atoms(Si4, ax, radii=0.3 , rotation=(xyz1), colors = ["green" ]*15 ) plot_atoms(Si5, ax, radii=0.3 , rotation=(xyz1), colors = ["blue" ]*15 ) plot_atoms(Si6, ax, radii=0.3 , rotation=(xyz1), colors = ["#7709f3" ]*15 ) ax.set_xlabel("x in $\mathrm{\AA}$" ) ax.set_ylabel("y in $\mathrm{\AA}$" ) handles, labels = ax.get_legend_handles_labels() red = mpatches.Patch(color='red' , label='Initial' ) orange = mpatches.Patch(color='orange' , label='Image 1' ) yellow = mpatches.Patch(color='yellow' , label='Image 2' ) green = mpatches.Patch(color='green' , label='Image 3' ) blue = mpatches.Patch(color='blue' , label='Image 4' ) purple = mpatches.Patch(color='#7709f3' , label='Final' ) ax.legend(handles = [red, orange, yellow, green, blue, purple], loc="upper right" ) fig.show()
1.7 cRPA 任务 输入文件 计算